Filgotinib programma in reuma
De reumatoïde artritis markt en de beperkingen van de huidige behandelingen
Reumatoïde artritis (reuma) is een chronische auto-immuunziekte gekenmerkt door ontsteking en degeneratie van de gewrichten. Het treft bijna 1% van de volwassen bevolking wereldwijd, manifesteert zich meestal tussen de leeftijden van 30 en 50 jaar en heeft een hoge prevalentie bij vrouwen. Patiënten lijden aan pijn, stijfheid en beperkte mobiliteit als gevolg van een hardnekkige ontsteking van meerdere gewrichten, wat uiteindelijk resulteert in onomkeerbare schade aan het gewrichtskraakbeen en bot. Naarmate reuma zich ontwikkelt, beschouwen de afweercellen van het immuunsysteem bepaalde lichaamseigen eiwitten alsof ze vreemd zijn en reageren de lymfocytcellen op deze eiwitten. De lymfocytreactie veroorzaakt dan het vrijkomen van cytokines, die chemische boodschappers zijn die tot meer ontstekingen en gewrichtsschade leiden. De ontsteking kan zich uitbreiden naar andere gebieden in het lichaam, waardoor uiteindelijk niet alleen gewrichtsschade, maar ook chronische pijn, vermoeidheid en verlies van functioneren veroorzaakt. De ontsteking kan ook leiden tot hart- en vaatziekten en een verhoogd risico op een hartaanval. Volgens het ACR (Amerikaans College van Reumatologie) verdubbelt het risico op een hartaanval binnen de eerste 10 jaar volgende op de diagnose van reuma.
De belangrijkste doelstellingen bij de behandeling van reuma zijn het inperken van de ontsteking en het vertragen of het stoppen van de progressie van de ziekte. Initiële therapeutische benaderingen waren gebaseerd op ziekte-modificerende anti-reumatische geneesmiddelen (DMARDS), zoals methotrexaat (MTX) en sulfasalazine. Deze oraal toegediende geneesmiddelen werken voornamelijk door het immuunsysteem te onderdrukken maar tegelijk zorgt een effectieve onderdrukking van het immuunsysteem voor een verhoogd risico op infecties. Deze medicijnen worden tevens geassocieerd met bijwerkingen zoals misselijkheid, buikpijn en ernstige long en lever toxiciteit. Bovendien, aangezien het gemiddeld 6-12 weken duurt vooraleer deze medicijnen werkzaam zijn, kan de reumatoloog de behandeling uitbreiden met vrij verkrijgbare pijnstillers of niet-steroïdale anti-inflammatoire geneesmiddelen (NSAIDs) om de pijn en ontsteking te behandelen. Ondanks deze tekortkomingen, worden DMARDS nog steeds beschouwd als de eerste lijn behandeling van reuma.
De ontwikkeling van monoklonale antilichamen en biologische geneesmiddelen betekende een significante vooruitgang in de behandeling van reuma. Therapieën met biologische geneesmiddelen omvatten het gebruik van antilichamen of andere eiwitten geproduceerd door levende organismen om een ziekte te behandelen. Bij sommige mensen met reuma is het TNF eiwit in grote hoeveelheden aanwezig in het bloed en in de gewrichten, wat ontsteking veroorzaakt samen met pijn en zwelling. Er werden therapieën met biologische geneesmiddelen ontwikkeld die de productie van TNF blokkeren door communicatie tussen de B-cellen van het immuunsysteem, die een rol spelen bij de pijn en zwelling veroorzaakt door artritis, te verstoren. Zulke anti-TNF’s zijn momenteel de standaard eerste en tweedelijns therapieën met biologische geneesmiddelen voor patiënten met reuma die een onvoldoende respons vertonen op behandeling met DMARDS. Aangezien anti-TNF middelen werkzaam zijn door onderdrukking van het immuunsysteem, leiden ze ook tot een aanzienlijke toename van het risico op infecties. Bovendien moeten alle goedgekeurde anti-TNF therapieën toegediend worden door injectie of intraveneuze injectie, wat onhandig en pijnlijk is voor sommige patiënten. Voor sommige patiënten die lijden aan gewrichtspijn en gewrichtsschade ten gevolge van reuma, kan het bijzonder moeilijk zijn deze geneesmiddelen toe te dienen door zelf-injectie.
Niet alle patiënten die met een anti-TNF behandeld worden, bereiken voldoende klinische respons of handhaven de bereikte klinische respons, waardoor de behoefte ontstaat om over te schakelen naar een nieuwe therapie ten einde de ziekte te bestrijden. Ongeveer een derde van de reuma patiënten reageert niet afdoende op een anti-TNF. Anti-TNF’s worden bovendien geassocieerd met een lage graad van ziekteremissie en de respons op deze middelen is meestal niet duurzaam. In meer dan 30% van deze populatie zijn alternatieve behandelingen nodig. Een aanzienlijk aantal patiënten behandeld met een anti-TNF therapie moet binnen 24 maanden een tweede en derde anti-TNF therapie proberen. Uit een prospectieve cohort studie van reuma-patiënten in Groot Brittannië bleek dat binnen de 15 maanden na de start van de behandeling met een anti-TNF, 12% van de patiënten overschakelden op een tweede anti-TNF therapie als gevolg van gebrek aan werkzaamheid, en 15% van de patiënten overschakelden op een tweede anti-TNF therapie wegens toxiciteit. Uiteindelijk moet 30% van de patiënten een alternatieve anti-TNF behandeling ondergaan. Het opeenvolgend overschakelen naar een andere anti-TNF therapie is een ernstig probleem voor patiënten. Aangezien het maanden kan duren voordat blijkt of een geneesmiddel werkt, draagt het opeenvolgend falen van therapie bij tot de progressie van de ziekte met voortschrijdende en onomkeerbare structurele gewrichtsbeschadiging tot gevolg. Voor reuma patiënten die niet op anti-TNF therapie reageren, of voor wie anti-TNF’s niet aangewezen zijn, zijn de orale JAK remmers en biologische geneesmiddelen met verschillende werkingsmechanismen in ontwikkeling.
2013 Reuma wereldwijde markt: $15,6 miljard
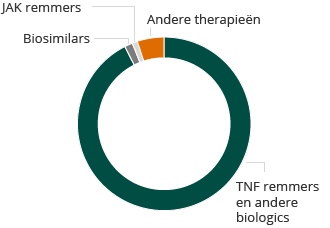

Ondanks deze beperkingen, is de wereldwijde markt voor reuma therapieën groot en groeit ze snel. De markt voor reuma therapieën in de 10 belangrijkste markten in de gezondheidszorg was $15,6 miljard in 2013 en zal naar verwachting groeien tot meer dan $19 miljard in 2023, volgens een GlobalData PharmaPoint rapport van december 2014. Injecteerbare biologische geneesmiddelen vormen het grootste deel van deze markt.
Ondanks de prevalentie van biologische geneesmiddelen voor de behandeling van reuma blijft er een grote medische nood aan geneesmiddelen met een betere werkzaamheid, en vooral een aanhoudende werkzaamheid, veiligheid en gemak van gebruik in vergelijking met de bestaande eerstelijns behandelingen.
Het potentieel van JAK remmers
De familie van JAK eiwitten bestaat uit vier tyrosine kinases, JAK1, JAK2, JAK3 en Tyk2 die betrokken zijn bij de JAK-gemedieerde signaaltransductie die normale hematopoëse (of het maken van bloed), ontsteking en het immuunsysteem reguleert. Ontregeling van de JAK signaaltransductie is geassocieerd met een aantal ziekten, waaronder reuma, psoriasis en andere chronische ontstekingsziekten. Daarom vormt de familie van JAK eiwitten sinds lang een interessegebied voor ontwikkelaars van nieuwe geneesmiddelen ter behandeling van zulke ziekten. Een groeiende hoeveelheid klinische gegevens suggereren dat de mate van selectiviteit van een potentieel nieuw geneesmiddel ten aanzien van de verschillende JAK eiwitten sterk samenhangt met het werkzaamheids- en veiligheidsprofiel van het potentieel geneesmiddel. Van JAK1 is bijvoorbeeld geweten dat het interageert met andere JAKs en zodoende onder andere cytokine gedreven pro-inflammatoire signaaltransductie stimuleert, wat leidt tot ontsteking in bepaalde menselijke weefsels. Bijgevolg wordt aangenomen dat de remming van JAK1 therapeutische mogelijkheden biedt ter behandeling van een scala van ontstekingsziekten en andere ziekten veroorzaakt door JAK-gemedieerde signaaltransductie. Remming van de andere drie tyrosine kinases uit de JAK familie daarentegen (JAK2, JAK3 en TYK2) is mogelijks niet noodzakelijk voor een anti-inflammatoire werking. Hun remming zou echter kunnen bijdragen tot bijwerkingen. Zo is gekend dat remming van JAK2 vaak gepaard gaat met bloedarmoede, terwijl remming van JAK3 leidt tot immunosuppressie. Van niet-selectieve JAK remmers is tevens aangetoond dat ze LDL cholesterol verhogen. Aldus wordt verondersteld dat het gewenste werkzaamheids- en veiligheidsprofiel van een JAK-remmer rechtstreeks in verband kan worden gebracht met de selectiviteit van het product.
In november 2012 werd het geneesmiddel Xeljanz door de Amerikaanse autoriteit FDA goedgekeurd voor commerciële verkoop als eerste en enige goedgekeurde JAK remmer voor de behandeling van reuma. Xeljanz is goedgekeurd voor de behandeling van volwassen patiënten met reuma die een ontoereikende klinische respons hebben gehad op, of die intolerant zijn, voor methotrexaat. Xeljanz is een klein molecuul geschikt voor orale toediening en heeft een sterke bindingsaffiniteit voor JAK3 en JAK1 en zwakkere affiniteit voor JAK2. De veiligheid en effectiviteit van Xeljanz werden geëvalueerd in zeven klinische studies bij volwassen patiënten met matig tot ernstig actieve reuma. In alle studies ondervonden patiënten behandeld met Xeljanz een verbetering van de klinische respons en fysiek functioneren vergeleken met patiënten behandeld met placebo. Echter, het gebruik van Xeljanz werd in verband gebracht met een reeks van bijwerkingen, waaronder bloedarmoede (verminderd hemoglobinegehalte) en verhogingen van zowel de hoeveelheden lever enzymen als cholesterolen in het bloed. Bijvoorbeeld, in gecontroleerde klinische studies met Xeljanz, werden dosisafhankelijke verhogingen in lipide parameters (totaal cholesterol, LDL-cholesterol, HDL-cholesterol, triglyceriden) waargenomen binnen de maand na de start van de behandeling. In de Xeljanz 5 mg tweemaal daags cohorte van de studie, werd een 15% toename in LDL-cholesterol vastgesteld, wat de goedgekeurde dosering in de Verenigde Staten betreft. Xeljanz werd door de Europese autoriteit EMA niet goedgekeurd voor commerciële verkoop in Europa. Onder andere hierdoor bestaat er nog steeds een grote behoefte aan nieuwe oraal toegediende geneesmiddelen ter behandeling van reuma en andere inflammatoire ziekten die een gunstiger profiel qua bijwerkingen hebben.
Galapagos’ filgotinib programma voor reuma
Door de hoge selectiviteit voor JAK1 bezit filgotinib, vergeleken met andere JAK remmers die minder selectief zijn voor JAK1, de mogelijkheid om een verbeterd profiel van bijwerkingen en verbeterde werkzaamheid bij reuma patiënten te bieden. Filgotinib wordt momenteel geëvalueerd in drie lopende Fase 2b studies, die gezamenlijk worden aangeduid als DARWIN, bij patiënten met matige tot ernstige reuma die een onvoldoende respons op eerstelijns behandeling methotrexaat lieten zien. Galapagos verwacht de resultaten na 12 weken behandeling in de DARWIN studies (DARWIN 1 en 2) in april 2015 en de definitieve resultaten na 24 weken behandeling in juli 2015. Daarnaast heeft Galapagos de DARWIN 3 studie lopen, dewelke een lange-termijn opvolgstudie is waarbij patiënten de mogelijkheid geboden wordt om op filgotinib behandeling te blijven. Van de patiënten die de DARWIN 1 of DARWIN 2 studie hebben doorlopen, heeft 98% van de in aanmerking komende patiënten gekozen voor deelname aan de DARWIN 3 opvolgstudie.
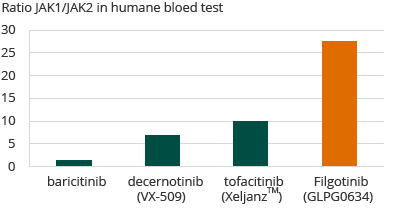
In een door Galapagos ontwikkelde menselijke bloed test, werd aangetoond dat filgotinib, in vergelijking met andere bekende JAK-remmers die ofwel zijn goedgekeurd voor verkoop of in klinische ontwikkeling zijn voor de behandeling van reuma, selectiever is voor JAK1. Filgotinib laat een 30-voudige selectiviteit voor JAK1 ten opzichte van JAK2 zien. Galapagos verwacht dat de hoge selectiviteit van filgotinib voor JAK1, ten opzichte van andere erkende reuma therapieën, de mogelijkheid biedt op een gelijkaardige of betere werkzaamheid terwijl het veiligheidsprofiel beter is als gevolg van de lage selectiviteit voor JAK2 en JAK3.
Selectiviteit van JAK remmers voor reuma

Bovendien biedt filgotinib de mogelijkheid om gebruikt te worden als een éénmaal daagse therapie, wat mogelijk tot een gemakkelijkere toedieningsmethode en hogere therapietrouw van patiënten leidt. Filgotinib heeft een profiel dat mogelijk veilig gebruik in combinatie met andere geneesmiddelen toelaat, hetgeen een belangrijke eigenschap is voor een behandeling in deze patiëntenpopulatie omdat de meeste van deze patiënten ook gelijktijdig op andere therapieën zitten.
Door het uitgebreide DARWIN klinische programma, heeft Galapagos tot doel om aan te tonen dat filgotinib zal voldoen aan de volgende klinische en producteigenschappen ter behandeling van reuma:
- Veiligheid: Dat filgotinib goed zal worden verdragen, niet tot behandeling geïnduceerde bloedarmoede, noch tot toename van de LDL/HDL balans zal leiden, en een lager totaal infectiepercentage zal hebben in vergelijking met andere goedgekeurde therapieën in reuma
- Werkzaamheid: Dat filgotinib een snelle aanvang van de werkzaamheid met een duurzame effect zal laten zien, gelijk aan of beter dan die van goedgekeurde biologische geneesmiddelen en therapieën zoals anti-TNF’s
- Gemak: Dat filgotinib een oraal, eenmaal daagse toediening zal toelaten
- Combinatie met andere therapieën: Dat filgotinib vanwege het ontbreken van negatieve wisselwerkingen met andere geneesmiddelen, veilig zal kunnen worden gecombineerd met andere therapieën die vaak voorgeschreven zijn bij reuma-patiënten
Filgotinib wordt momenteel geëvalueerd in drie lopende Fase 2b studies bij patiënten met matige tot ernstige reuma en die een onvoldoende respons op methotrexaat hebben getoond. DARWIN 1 en DARWIN 2 zijn studies die als doel hebben de gepaste dosis te bepalen. DARWIN 3 is een lange-termijn opvolgstudie die het mogelijk maakt dat patiënten van de DARWIN 1 en 2 studies doorgaan met de filgotinib behandeling. Deze wereldwijde Fase 2b studies zijn volledig gerekruteerd. De eerste resultaten na 12 weken behandeling in de DARWIN 1 studie zullen naar verwachting in april 2015 beschikbaar zijn en de definitieve resultaten na 24 weken behandeling voor deze studie zullen naar verwachting in juli 2015 bekend zijn.
Galapagos heeft een exclusieve samenwerkingsovereenkomst met AbbVie om filgotinib te ontwikkelen en te commercialiseren. In het kader van deze overeenkomst, is Galapagos verantwoordelijk voor de voortgang van de drie Fase 2 studies bij reuma en de ziekte van Crohn. Als AbbVie bepaalt dat de resultaten na 24 weken van behandeling van de eerste twee van deze studies (DARWIN 1 en 2) aan een aantal criteria voldoen, zal AbbVie geacht worden een licentie te hebben genomen. Als de opgegeven criteria niet worden behaald, heeft AbbVie de mogelijkheid om voor een licentie te kiezen nadat de 24 weken resultaten uit deze studies bekend zijn. Mocht AbbVie een licentie nemen, zal AbbVie alleen verantwoordelijk zijn voor de Fase 3 klinische ontwikkeling, de wereldwijde productie en commercialisering van filgotinib. Galapagos behoudt een optie op bepaalde co-promotie rechten in Nederland, België en Luxemburg, en komt in aanmerking voor potentiële toekomstige succesbetalingen en royalty’s op de eventuele wereldwijde commerciële verkoop in alle goedgekeurde indicaties voor filgotinib.