Galapagos programs in cystic fibrosis
The unmet need in CF
CF is an area of significant unmet medical need for which Galapagos is developing a three-product combination therapy.
CF is a rare, life-threatening, genetic disease that affects approximately 80,000 patients worldwide. CF is a chronic disease that affects the lungs and digestive system. CF patients, with significantly impaired quality of life, have an average lifespan approximately 50% shorter than the population average, with the median age of death at 37. There currently is no cure for CF. CF patients require lifelong treatment with multiple daily medications, frequent hospitalizations and ultimately lung transplant, which is life-extending but not curative. In the United States, a CF patient on average incurs approximately $50,000 per year, or $1,350,000 over his or her lifetime, in outpatient expenses alone and substantial additional costs for frequent hospitalizations. Kalydeco, the only approved therapy for the underlying cause of CF, adds approximately $300,000 of additional costs per year.
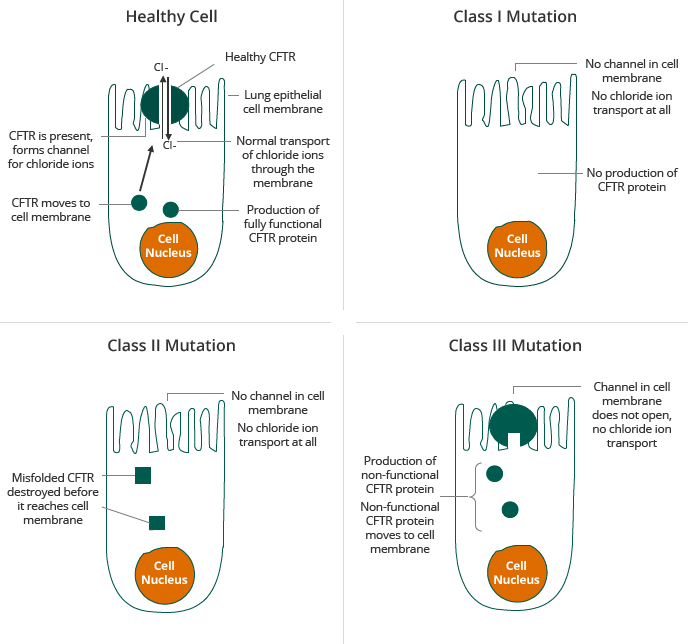
CF is caused by a mutation in the gene for the CFTR protein, which results in abnormal transport of chloride across cell membranes. Transport of chloride is required for effective hydration of epithelial surfaces in many organs of the body. Normal CFTR channel moves chloride ions to outside of the cell. Mutant CFTR channel does not move chloride ions, causing sticky mucous to build up on the outside of the cell. CFTR dysfunction results in dehydration of dependent epithelial surfaces, leading to damage of the affected tissues and subsequent disease, such as lung disease, malabsorption in the intestinal tract and pancreatic insufficiency.
Individuals who carry two copies of a defective CFTR gene, referred to as homozygous, are typically affected by CF and show symptoms of the disease. Individuals who carry one copy of a defective CFTR gene are called carriers. Carriers are typically unaffected by CF and show no symptoms of the disease. Individuals who carry one copy each of two different defective CFTR genes are referred to as heterozygous. They are typically affected by CF and show symptoms of the disease. Today, the majority of CF patients are diagnosed at birth through newborn screening and approximately 92% of diagnosed patients have been genotyped. There are more than 1,900 known mutations in the CFTR gene. Mutations in the CFTR gene can be classified into six classes according the mode by which they disrupt the synthesis, traffic and function of CFTR, as described in the diagram below.
CF Mutations

Source: Adapted from [Proesmans et al., 2008]
The two most prevalent mutations in the CFTR gene are Class II and Class III, including the F508del mutation and the G551D mutation, respectively. In Class II patients, insufficient CFTR reaches the membrane, about 50% of the patients have the F508del mutation on both alleles, the so-called homozygotes. For clinical trials, these patients form a homogenous group. The other 50% of the patients, have the F508del mutation on one allele only and carry another mutation on the second allele, they are called the heterozygotes. Also this other mutation impairs the correct processing of CFTR. As the group is less homogenous, clinical trials have proven to be more difficult. The F508del mutation is sometimes called a “processing” mutation because it results in a defect in the CFTR protein in which the CFTR protein does not reach the surface of cells in sufficient quantities. The G551D mutation, a Class III mutation, is sometimes called a “gating” mutation because it results in a defect in the CFTR protein in which the defective CFTR protein reaches the surface of a cell but does not efficiently transport chloride ions across the cell membrane. Most therapeutic approaches under development for CF target the defects caused by one or both of these mutations. Given the prevalence of the F508del mutation, a compound that corrects the effect of the F508del mutation can, beside for patients with Class II mutations only, also be used for combination therapy approaches in heterozygous patients with Class I and Class III mutations.
The potential of CFTR modulators (potentiators and correctors) for the treatment of CF
There is no cure for CF, and to date, all but one of the therapies approved to treat CF patients have been designed to treat the symptoms rather than address the underlying cause of the disease. The market for CF therapies, across the six main healthcare markets, exceeded $1 billion in 2012 and is to exceed $5 billion in 2018 according to a July 2014 GlobalData OpportunityAnalyzer report, primarily driven by introduction of disease modifying treatments. To treat the symptoms of disease, such as CF-associated malnutrition, diabetes, lung disease and systemic inflammation, an aggressive combination of specific therapies is required. To address the cause of the disease, the primary focus has been on a class of drugs known as CFTR modulators.
Kalydeco, marketed by Vertex, is currently the only approved therapy to address the cause of CF. Kalydeco is an orally-administered CFTR potentiator for the treatment of patients two years of age and older with CF who have the Class III (G551D) gating mutation in their CFTR gene. Kalydeco is designed to keep the CFTR protein channels on the cell surface open longer in order to increase the flow of salt and water into and out of the cell. However, this treatment is limited to the subset of patients who suffer from the Class III and other gating mutations of the CFTR gene. Class III mutations occur in only a small percentage of patients with CF (3%).
In contrast, the Class II F508del mutation affects close to 90% of all CF patients. In these patients, CFTR is not expressed at the cell surface and cannot be potentiated by drugs like Kalydeco (that can only function if CFTR is already present in the cell membrane). Small molecule corrector approaches aim to transport the non-functional Class II CFTR protein to the cell membrane. Other companies currently developing small molecule correctors include Vertex, Pfizer, Flatley Laboratories, Genzyme, Targeted Genetics and Bayer. To date, however, there are no approved corrector molecules on the market.
The Class I mutations affect approximately 7% of all CF patients. This mutation shortens the length of the CFTR protein and leads to complete loss of CFTR function. To date, there are no approved molecules on the market to treat this mutation.
Lumacaftor (VX-809), which is being developed by Vertex, is a small molecule corrector being studied in patients with two copies (homozygous) of the Class II (F508del) mutation in their CFTR gene for use in combination with Kalydeco. In June 2014, Vertex announced that its two Phase 3 clinical trials of lumacaftor, when used in combination with Kalydeco in CF patients homozygous for the Class II (F508del) mutation, showed statistically significant improvement in the trial’s primary endpoint of improved lung function, compared to placebo. Vertex also showed statistically significant reductions in pulmonary exacerbations in the pooled analysis of both studies. Other signs of clinical improvement were either limited or not statistically different from placebo.
Despite the approval of Kalydeco and the pending approval of Kalydeco/lumacaftor combinations, there is need for better therapies with improved pulmonary function. Though many pediatric patients have normal lung function at the time of diagnosis, physicians generally believe that earlier treatments can have downstream benefits for the patient by slowing the deterioration in lung function.
Galapagos believes that restoration of CFTR function in cellular assays may be predictive of clinical outcomes. Specifically, review of Vertex patient and cellular data has shown strong correlation as reflected in the diagram below.
F508del – Homozygous for F508del
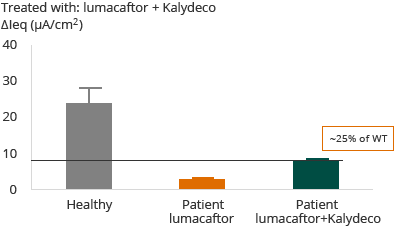

In the case of patients with F508del mutation, the administration of Kalydeco and lumacaftor combination resulted in approximately 20% restoration of normal, or wild-type, CFTR. The clinical outcome reflected in Vertex’s Phase 3 trial and primary endpoint was that 46% of patients showed an FEV1 improvement of greater than or equal to 5%. Forced expiratory volume (FEV1) levels are a measurement of the volume of air that can be forcibly blown out in one second after full inspiration.
G551D – Heterozygous G551D with F508del
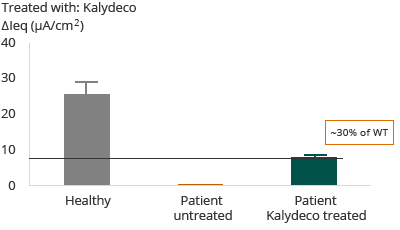

Further, as reflected in the diagram above, for patients with G551D mutation, the administration of Kalydeco resulted in approximately 30% restoration of wild-type CFTR. The clinical outcome reflected in Vertex’ Phase 3 trial and primary endpoint was that 75% of patients showed an FEV1 improvement of greater than or equal to 5%.
Galapagos believes these studies demonstrate that cellular models can be used to identify novel molecules to treat Class II and Class III mutations and select those combinations that can restore wild-type CFTR to greater than 50%, a threshold that may need to be achieved to lead to disease remission in patients.
Galapagos’ programs in CF
Galapagos has an exclusive collaboration agreement with AbbVie to discover, develop and commercialize novel CF modulators. AbbVie and Galapagos are working collaboratively, contributing technologies and resources to develop and commercialize oral drugs that address the main mutations in CF patients, including Class II and Class III.
Galapagos’ CF modulators may have the potential to offer important advantages compared to currently approved therapies as well as other therapies under development:
- disease modifying activity in Class II/III mutations in CF
- regaining greater than 50% of CFTR activity, important for achieving compelling clinical efficacy
- improved risk/benefit compared to standard of care
- small molecules allowing for oral administration
- adequate safety for chronic use, including pediatric application
- no adverse interactions with drugs commonly taken by CF patients, including antibiotics and anti-inflammatory drugs
- effective in homo- & heterozygous patients
Galapagos may be well positioned in CF due to its:
- robust portfolio of CF modulators, including prolific chemistry with multiple binding modes to modulate CFTR
- unique assay cascade, including primary cells from CF patients, for screening of candidate drugs that modulate the CFTR protein
- expertise in working since 2008 with a broad discovery platform containing highly relevant disease assays starting from cells from CF patients
- collaborative partnership with AbbVie, which is an expert in combination therapies and committed to the CF field
Galapagos novel modulator combinations for treating CF
Galapagos is developing novel oral corrector-potentiator combinations for the treatment of CF patients with the Class II F508del mutation, including both homozygous and heterozygous patients. The aim is to develop multiple correctors and multiple potentiators for patients with this mutation, and Galapagos has been successful in identifying multiple candidates in each focus area thus far.
Therapies that restore CFTR function through a combination of correctors and potentiators improve hydration of the lung surface and subsequent restoration of mucociliary clearance. Galapagos is focused on increasing the percentage of wild-type CFTR restored to greater than 50%. A potentiator/corrector combination restoring more than 50% of healthy function CFTR may have a substantially positive impact on the quality of life of Class II patients and may reverse disease.
Galapagos has identified multiple series of novel corrector molecules that enhance the restoration of CFTR in combination with novel potentiator GLPG1837. Based on pre-clinical data, potentiator GLPG1837 may have the potential to offer a superior efficacy and safety profile compared to Kalydeco, important for Class III positioning, but also important for forming the potentiator component of superior combination therapies for Class II mutation patients as well. As reflected below, Galapagos’ triple combination therapy of GLPG1837 plus corrector candidate GLPG2222 plus other molecules from our other corrector series show up to 60% restoration of wild-type CFTR function in pre-clinical tests, compared to the 20% demonstrated by the Kalydeco/lumacaftor combination. In addition, Galapagos’ triple combination therapy could offer the ability to combine with antibiotics and other therapies often prescribed to CF patients, and these molecules appear to have the potential for no drug-drug interaction liabilities, important for CF patients who use multiple medications like antibiotics.
The diagram below is a pre-clinical evaluation of Class II homozygous primary cells.
Galapagos in-house pre-clinical evaluation of various compounds in lung epithelial cells from Class II mutation patients
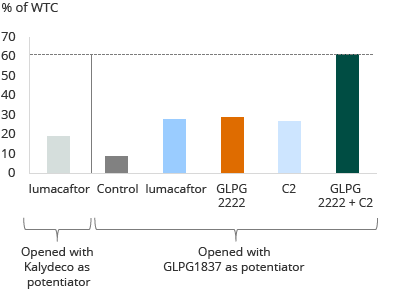

Lumacaftor and Kalydeco achieve approximately 20% wild-type restoration on average in this assay. The other bars show potentiator GLPG1837 in combination with lumacaftor, with GLPG2222, and with another corrector candidate, or a combination of GLPG2222 and another corrector candidate, all tested in this assay in the same donor cells. These compounds may make a clinical difference for heterozygous Class II patients. Based on pre-clinical data, potentiator GLPG1837, in combination with GLPG2222 plus other molecules from the C2 corrector series showed a 60% restoration of wild-type, as shown in the diagram above.