Filgotinib program in rheumatoid arthritis
The RA market and limitations of current treatments
RA is a chronic autoimmune disease, characterized by inflammation and degeneration of the joints. It affects almost 1% of the adult population worldwide, with onset typically between the ages of 30 and 50 years, and with a high prevalence in women. Patients suffer from pain, stiffness, and restricted mobility due to a persistent inflammation of multiple joints, which ultimately results in irreversible damage of the joint cartilage and bone. As RA develops, the body’s immune cells perceive the body’s own protein as foreign and cells called lymphocytes react to this protein. The reaction then causes the release of cytokines, which are chemical messengers that trigger more inflammation and joint damage. The inflammation may spread to other areas in the body, ultimately causing not only joint damage but also chronic pain, fatigue, and loss of function. Inflammation has also been linked to heart disease and the risk of having a heart attack. RA nearly doubles the risk of having a heart attack within the first 10 years of being diagnosed, according to the ACR.
The primary goals in the treatment of RA are to control inflammation and slow or stop disease progression. Initial therapeutic approaches relied on disease-modifying anti-rheumatic drugs, or DMARDS, such as methotrexate and sulphasalazine. These oral drugs work primarily to suppress the immune system and, while effective in this regard, the suppression of the immune system leads to an increased risk of infections. These drugs are also associated with side effects including nausea, abdominal pain, and serious lung and liver toxicities. Further, because these drugs often take on average from 6-12 weeks to take effect, rheumatologists may also couple them with over-the-counter pain medications or non-steroidal anti-inflammatory drugs, or NSAIDs, to treat the pain and inflammation. Despite their serious shortcomings, DMARDS are still considered first-line therapies.
The development of monoclonal antibodies and biologics represented a significant advance in RA treatment. Biologic therapies involve the use of antibodies or other proteins produced by living organisms to treat disease. In some people with arthritis, the TNF protein is present in the blood and joints in excessive amounts, thereby increasing inflammation, along with pain and swelling. Biologic therapies have been developed to address this overproduction of TNF by disrupting communication between the body’s immune cells. Thus, they block the production of TNF or are designed to attach to and destroy the body’s immune B-cells, which play a part in the pain and swelling caused by arthritis. Anti-TNFs are currently the standard of care for first- and second-line biologic therapies for RA patients who have an inadequate response to DMARDS. Since anti-TNF drugs function through a suppression of the immune system, they also lead to a significant increase in the risk of infections. In addition, all approved anti-TNFs need to be delivered by injection or intravenously, which is inconvenient and painful for some patients, and in some cases self-injection can be particularly difficult for patients who suffer joint pain and damage from RA.
Not all patients achieve sufficient clinical response or maintain clinical response to anti-TNFs over time, resulting in a need to switch or cycle to a new therapy to control their disease. Approximately one-third of RA patients do not adequately respond to anti-TNFs. In addition, anti-TNFs are associated with low rates of disease remission and the response to these agents is not typically durable. In more than 30% of this population, alternative treatment approaches are needed. A significant number of patients treated with an anti-TNF will be cycled to their second and third anti-TNF within 24 months of anti-TNF therapy initiation. A prospective cohort study of RA patients from a UK national register of new anti-TNF treatments showed that, within 15 months of treatment, 12% cycle to a second anti-TNF due to inefficacy, and 15% cycle to a second anti-TNF due to toxicity. Ultimately, 30% of patients need an alternative to anti-TNF treatment. Therapeutic cycling is a serious issue for patients because the efficacy of each successive drug is not known typically for several months, which contributes to progression of disease and continued irreversible structural joint damage. For RA patients who fail or for whom anti-TNFs are contraindicated, the oral agent JAK inhibitors and biologics with distinct mechanisms are in development.
2013 RA Worldwide Market: $15.6B
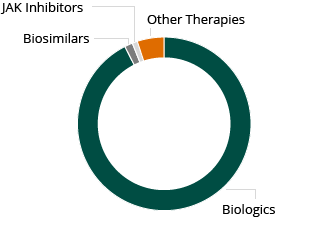

Despite these limitations, the global market for RA therapies is large and growing rapidly. The market for RA therapies across the 10 main healthcare markets was $15.6 billion in 2013 and is expected to grow in excess of $19 billion by 2023, according to a December 2014 GlobalData PharmaPoint report. Injectable, biological therapies are the largest component of this market.
However, despite the prevalence of biologics in the treatment of RA, there continues to be a considerable unmet need with regard to efficacy, including sustained efficacy, safety, and convenience of use with these existing first line treatments.
The potential of JAK inhibitors
The family of JAKs is composed of four tyrosine kinases, JAK1, JAK2, JAK3 and Tyk2 that are involved in the JAK signaling pathway, which regulates normal hematopoiesis, or blood making, inflammation and immune function. Dysregulation of the JAK signaling pathway has been associated with a number of diseases, including RA, psoriasis and other chronic inflammatory diseases. Accordingly, the JAK family has long been an area of interest for drug developers working in these areas.
A growing body of clinical data suggests that the level of selectivity of a JAK therapeutic is highly correlated to its efficacy and safety profile. For example, JAK1 is known to interact with the other JAKs, among others, to transduce cytokine-driven pro-inflammatory signaling, which leads to inflammation in human tissues. Therefore, inhibition of JAK1 is believed to be of therapeutic benefit for a range of inflammatory conditions as well as for other diseases driven by JAK-mediated signal transduction. In contrast, inhibition of the other three kinases (JAK2, JAK3, and TYK2) may not be required for the anti-inflammatory effect, whereas their inhibition may contribute to side effects. For example, inhibition of JAK2 has been linked to anemia, and inhibition of JAK3 to immunosuppression. Non-selective JAK inhibitors have been shown to increase LDL cholesterol. Therefore, the desired efficacy and safety profile of any JAK inhibitor may be directly linked to the selectivity of the product.
In November 2012, Xeljanz was approved by the FDA as the first and only JAK inhibitor for RA approved for commercial sale in the United States. Xeljanz is intended for the treatment of adult patients with RA who have had an inadequate response to, or who are intolerant of, methotrexate. Xeljanz is a small molecule suitable for oral administration and has strong binding affinity for JAK3 and JAK1, and weaker affinity for JAK2. The safety and effectiveness of Xeljanz were evaluated in seven clinical trials in adult patients with moderately to severely active RA. In all of the trials, patients treated with Xeljanz experienced improvement in clinical response and physical functioning compared to patients treated with placebo. However, the use of Xeljanz has been associated with a range of side effects, including anemia (reduced hemoglobin levels) and elevations in both liver enzyme and lipid levels. For example, in controlled clinical trials for Xeljanz, dose-related elevations in lipid parameters (total cholesterol, LDL cholesterol, HDL cholesterol, triglycerides) were observed at one month of exposure, including a 15% increase in LDL cholesterol in the Xeljanz 5 mg twice daily arm, the approved dosage in the United States. Xeljanz was not approved in Europe. Accordingly, there continues to be a significant unmet medical need in RA and other inflammatory diseases for an orally administered approach with a more favorable side effect profile.
Galapagos’ filgotinib program for RA
Due to its high selectivity for JAK1, filgotinib may have the potential to offer an improved side effect profile and improved efficacy in RA patients as compared to other JAK inhibitors which are less selective for JAK1. Filgotinib is currently being evaluated in three ongoing Phase 2b trials, which are referred to collectively as DARWIN, in patients with moderate to severe RA who have an inadequate response to methotrexate (MTX) a common first line treatment for RA. Galapagos expects topline results from 12 weeks of treatment in the DARWIN trials (DARWIN 1 and 2) in April 2015 and final results from 24 weeks of treatment in July 2015. In addition, Galapagos is conducting DARWIN 3, a long-term follow-up trial that allows patients to remain on filgotinib treatment. Of the patients who have completed DARWIN 1 and DARWIN 2, 98% of eligible patients has elected to participate in the DARWIN 3 follow-up trial.
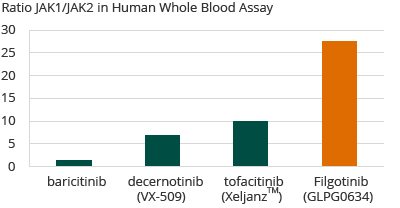
In an in-house human whole blood assay, Galapagos demonstrated that filgotinib was more selective for JAK1 than any other known compound that is either approved for sale or in clinical development, with a 30-fold selectivity for JAK1 over JAK2. Galapagos anticipates that the high selectivity of filgotinib for JAK1 may allow for efficacy equal to or better than that of other approved RA therapies, with an improved safety profile due to less selectivity for JAK2 and JAK3.
Selectivity of JAK Inhibitors in RA

Moreover, filgotinib may have the potential to be used as a once-daily therapy, thereby potentially improving ease of administration and patient compliance. Filgotinib has the potential to be used safely with concomitant medications, an important feature for this patient population since many of these patients are on other therapies to address co-morbities or other diseases.
Through the extensive DARWIN clinical programs, Galapagos aims to demonstrate the following clinical and product benefits of filgotinib for the treatment of RA:
- Safety: That filgotinib will be well tolerated, will show absence of treatment-induced anemia, will show no increase of LDL/HDL balance and will result in an overall lower infection rate as compared to other approved RA therapies.
- Efficacy: That filgotinib will enable rapid onset of action with durable efficacy equal to or better than approved biologics and approaches such as anti-TNFs.
- Convenience: That filgotinib will enable oral, once-daily dosing.
- Combination with other therapies: That filgotinib will be able to be safely combined with other therapies commonly prescribed to RA patients, due to its lack of drug-drug interactions.
Filgotinib is currently being evaluated in three ongoing Phase 2b trials in patients with moderate to severe RA and who have demonstrated an inadequate response to MTX. DARWIN 1 and DARWIN 2 are dose finding trials. DARWIN 3 is a long-term follow-up trial that allows patients to roll-over from DARWIN 1 and 2 trials and remain on treatment. These global Phase 2b trials are fully recruited. Topline results after 12 weeks of treatment in the DARWIN studies are expected in April 2015 and final results after 24 weeks of treatment for these studies are expected in July 2015.
Galapagos has an exclusive collaboration agreement with AbbVie to develop and commercialize filgotinib. Under this agreement, Galapagos is responsible for the advancement of three Phase 2 trials in RA and CD. If AbbVie determines that the first two of these trials (DARWIN 1 and 2) meet certain specified criteria, AbbVie will be deemed to have in-licensed the compound. If the specified criteria are not met, AbbVie has the opportunity to elect to in-license the compound following our delivery of the final data package from these trials. Should AbbVie in-license these programs, AbbVie will assume sole responsibility for Phase 3 clinical development, global manufacturing and commercialization of filgotinib. Galapagos retains an option to exercise certain co-promotion rights in the Netherlands, Belgium and Luxembourg, and Galapagos will be entitled to potential future regulatory and commercial milestone payments and royalties on global commercial sales across all approved indications for this compound, if any.