Our third treatment area is CF: an area of significant unmet medical need
Cystic fibrosis is a rare, life-threatening, genetic disease that affects approximately 80,000 patients worldwide and approximately 30,000 patients in the United States. CF is a chronic disease that affects the lungs and digestive system. CF patients, with significantly impaired quality of life, have an average lifespan approximately 50% shorter than the population average. There currently is no cure for CF. CF patients require lifelong treatment with multiple daily medications, frequent hospitalizations and ultimately lung transplant, which is life-extending but not curative. In the United States, a CF patient on average incurs approximately $50,000 per year in outpatient expenses alone and substantial additional costs for frequent hospitalizations. Kalydeco, the only approved therapy for the underlying cause of Class III mutation CF, adds approximately $300,000 of additional costs per year. Orkambi, the only approved therapy for the underlying cause of Class II mutation CF, adds approximately $259,000 of additional costs per year.
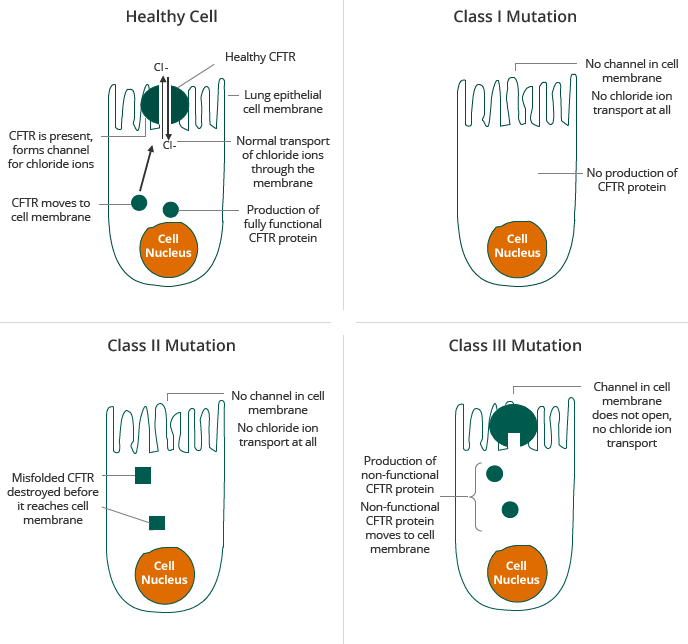
CF is caused by a mutation in the gene for the CF transmembrane regulator, or CFTR, protein, which results in abnormal transport of chloride across cell membranes. Transport of chloride is required for effective hydration of epithelial surfaces in many organs of the body. Normal CFTR channel moves chloride ions to the outside of the cell. Mutant CFTR channel does not move chloride ions, causing sticky mucous to build up on the outside of the cell. CFTR dysfunction results in dehydration of dependent epithelial surfaces, leading to damage of the affected tissues and subsequent disease, such as lung disease, malabsorption in the intestinal tract and pancreatic insufficiency.
Individuals who carry two copies of a defective CFTR gene are typically affected by CF and show symptoms of the disease. Individuals who carry one copy of a defective CFTR gene are called carriers. Carriers are typically unaffected by CF and show no symptoms of the disease. Individuals who carry one copy each of two different defective CFTR genes, referred to as heterozygous, are typically affected by CF and show symptoms of the disease. Today, the majority of CF patients are diagnosed at birth through newborn screening and the majority of diagnosed patients have been genotyped, up to 97% in the United States. There are more than 1,900 known mutations in the CFTR gene, some of which result in CF. Mutations in the CFTR gene can be classified into five classes according the mode by which they disrupt the synthesis, traffic and function of CFTR, as described in the table below.
Class |
CFTR Dysfunction |
CFTR Impact |
Commentary |
|
|---|---|---|---|---|
I |
Absent functional CFTR |
Protein translation |
Leads to no CFTR on cell membrane |
"Severe" Mutations ~96% of patients |
II |
Absent function CFTR |
Protein folding |
CFTR can't reach cell surface (F508del most common Class II) |
|
III |
Defective channel regulation |
Function |
CFTR on cell surface but can't be activated (G551D most common Class III) |
|
IV |
Defective CFTR channel |
Function |
CFTR on cell surface but chloride channel is unable to function properly |
"Mild" Mutations |
V |
Reduced function & synthesis |
Reduced number & CFTR degradation |
CFTR made at insufficient levels or degrades too quickly |
|
Selected CF Mutations

Source: Adapted from [Proesmans et al., 2008]
Two of the most prevalent mutations in the CFTR gene are Class II and Class III, including the F508del mutation and the G551D mutation, respectively. In Class II patients having insufficient CFTR reaching the membrane, about half of the patient population have the F508del mutation on both alleles, the so-called homozygotes. For clinical trials, these patients form a homogenous group. About the other half of the Class II patient population have the F508del mutation on one allele only and carry another mutation on the second allele; they are called the heterozygotes. Also this other mutation impairs the correct processing of CFTR. As the group is less homogenous, clinical trials have proven to be more difficult. The F508del mutation is sometimes called a “processing” mutation because it results in a defect in the CFTR protein in which the CFTR protein does not reach the surface of cells in sufficient quantities. The G551D mutation, a Class III mutation, is sometimes called a “gating” mutation because it results in a defect in the CFTR protein in which the defective CFTR protein reaches the surface of a cell but does not efficiently transport chloride ions across the cell membrane. Most therapeutic approaches under development for CF target the defects caused by one or both of these mutations. Given the prevalence of the F508del mutation, a compound that corrects the effect of the F508del mutation can, beside for patients with Class II mutations only, also be used for combination therapy approaches in heterozygous patients with Class I and Class III mutations.