Our CF program
CF is a rare, life-threatening, genetic disease affecting the lungs and the digestive system, impacting approximately 80,000 patients worldwide with approximately 30,000 patients in the United States. CF patients carry a defective cystic fibrosis transmembrane conductance regulator, or CFTR, gene and are classified based on their specific mutation of the CFTR gene. The Class II mutation is present in approximately 90% of CF patients, with Orkambi®1Orkambi is marketed by Vertex Pharmaceuticals being the only approved therapy for the underlying cause of CF in this mutation. Kalydeco is a disease-modifying treatment for Class III mutations, representing 4% of total CF patients. The market for CF therapies is robust and growing. According to Vertex Pharmaceuticals, approximately 9,000 patients were treated with Vertex therapies in 2016, and this they expect to grow to approximately 75,000 patients by 2024. Combined sales of Kalydeco and Orkambi were approximately $1.7 billion in 2016.
Despite the approval of Kalydeco and Orkambi, there is need for better therapies to improve pulmonary function for the majority of the patient population. Though many pediatric patients have normal lung function at the time of diagnosis, physicians generally believe that earlier treatments can have downstream benefits for the patient by slowing the deterioration in lung function.
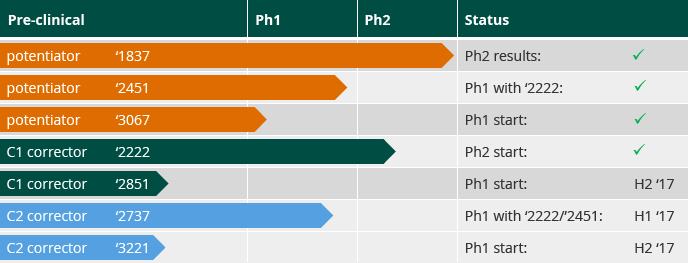
Two types of disease-modifying CFTR modulators have the current focus of CF drug developers. Potentiator molecules aim to restore the flow of ions through an activated CFTR by influencing the channel’s opening. Corrector molecules aim to overcome defective protein processing by restoring proper folding of CFTR and allowing for increased cell surface expression. In order to improve CFTR function meaningfully for the largest patient group with Class II, and Class III/IV, and other mutations, we believe a combination of medicines will be required, comprising a potentiator and two novel corrector (which we refer to as C1 and C2) molecules.
In pre-clinical cellular assay studies, we consistently observed that combinations of potentiator, C1, and C2 correctors restore close to healthy CFTR function in lung epithelial cells and organoids cultured from Class II patients, both homozygous and heterozygous for F508del, respectively:
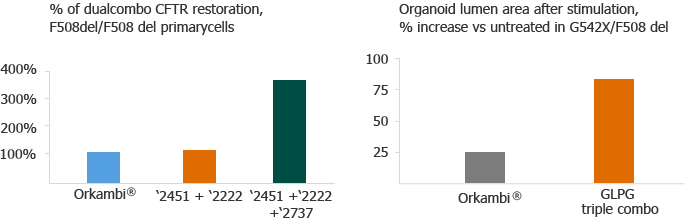

These results are suggestive of a compelling therapeutic option for these patients. We believe that our CF combination therapy may address the unmet need in both homozygous and heterozygous Class II patients, based on these in vitro results.
We aim to evaluate a once-daily, oral, triple combination CF therapy in patients starting in mid-2017, with additional trials with novel CF compounds initiating throughout 2017. We have developed a portfolio of lead and follow-on compounds from which to select the best potentiator and corrector molecules for our triple combination therapy: