Ons derde indicatiegebied is CF: een ziekte met een belangrijke onvervulde medische nood
Cystic fibrosis (CF) of taaislijmziekte is een zeldzame, levensbedreigende, genetische ziekte die de longen en het spijsverteringssysteem aantast. Over de hele wereld zijn er 80.000 mensen met CF, waarvan ongeveer 30.000 in de Verenigde Staten. Het is een chronische ziekte die voornamelijk de longen en het spijsverteringsstelsel aantast. Patiënten met CF hebben een ernstig verminderde kwaliteit van leven en gemiddeld een gehalveerde levensduur vergeleken met die van de gemiddelde bevolking. Momenteel is er geen remedie voor deze ziekte. Patiënten ondergaan een levenslange behandeling met meerdere dagelijkse medicaties, frequente hospitalisaties en uiteindelijk longtransplantatie. Deze symptomatische behandelingen zijn levensverlengend maar niet curatief. In de Verenigde Staten kost een gemiddelde behandeling van één patiënt ongeveer $50.000 per jaar aan ambulante kosten. Daarbovenop komen nog aanzienlijke extra kosten voor frequente hospitalisaties. Kalydeco, de enige goedgekeurde therapie voor een onderliggende oorzaak van Klasse III CF, voegt ongeveer $300.000 toe aan additionele kosten per patiënt per jaar. Door Orkambi, de enige goedgekeurde therapie voor de onderliggende oorzaak van CF met Klasse II-mutaties, worden jaarlijks circa $259.000 aan extra kosten toegevoegd.
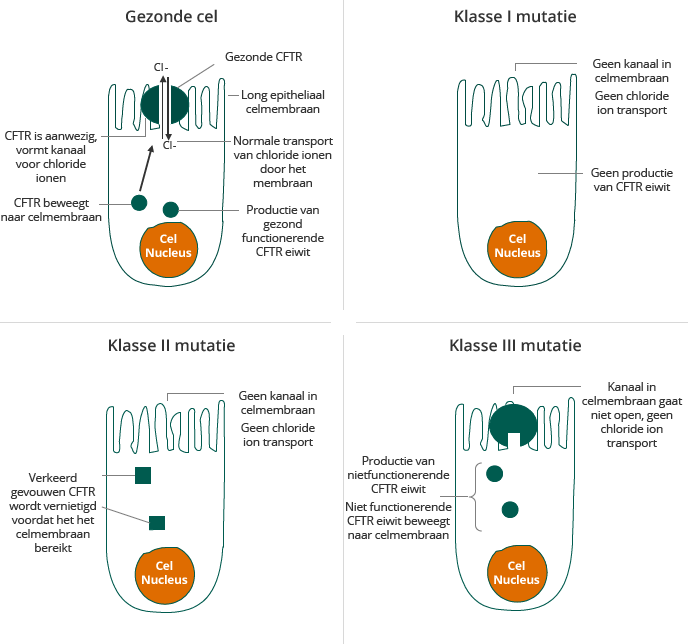
CF wordt veroorzaakt door een mutatie in het gen coderend voor het CFTR-eiwit, wat resulteert in abnormaal transport van chloride over het celmembraan. Het chloride transport is vereist voor een effectieve hydratatie van de epitheliale oppervlakken in vele organen van het lichaam. Een normaal CFTR kanaal zorgt voor de uitscheiding van chloride-ionen. Een gemuteerd CFTR kanaal heeft geen chloride transport, waardoor weinig water wordt aangetrokken of vastgehouden en een taai slijm ophoopt op de buitenkant van de cel. CFTR dysfunctie veroorzaakt dehydratatie van de epitheliale oppervlakken waardoor weefsels aangetast worden en dit vervolgens leidt tot ziekte van de longen, slechte absorptie in het darmkanaal en alvleesklier insufficiëntie.
Personen die twee kopieën van hetzelfde defect in het CFTR-gen dragen lijden normaliter aan CF en vertonen symptomen. Personen die één kopie van een defect CFTR-gen dragen en een gezond CFTR-gen op het tweede allel zijn dragers van de ziekte. Dragers lijden niet aan CF en vertonen geen symptomen. Tegenwoordig wordt de ziekte snel vastgesteld door het screenen van pasgeborenen en de meerderheid (ongeveer 97% in de V.S.) van de gediagnosticeerde patiënten worden tevens gegenotypeerd. Er zijn meer dan 1.900 bekende mutaties in het CFTR-gen. Mutaties in het CFTR-gen kunnen worden ingedeeld in zes klassen gebaseerd op de wijze waarop ze de synthese, vouwing, transport en functie van CFTR verstoren, zoals beschreven in de onderstaande tabel.
|
Klasse |
CFTR afwijking |
Impact op CFTR |
Commentaar | |
|---|---|---|---|---|
|
I |
Afwezigheid functioneel CFTR |
Eiwit translatie |
Leidt tot afwezigheid van CFTR op het celmembraan |
"Ernstige" Mutaties ~96% van de patiënten |
|
II |
Afwezigheid functioneel CFTR |
Vouwing van het eiwit |
CFTR kan het celmembraan niet bereiken (F508del meest voorkomend Klasse II) |
|
|
III |
Defecte kanaal regulatie |
Functie |
CFTR op celmembraan, maar kan niet worden geactiveerd (G551D meest voorkomend Klasse III) |
|
|
IV |
Defect CFTR kanaal |
Functie |
CFTR op celmembraan, maar het chloridekanaal kan niet correct functioneren |
"Milde" Mutaties |
|
V |
Verminderde functie & synthese |
Verminderd aantal & degradatie van CFTR |
Onvoldoende productie van CFTR of CFTR degradeert te snel |
|
CF mutaties

Bron: Galapagos, aangepast van Proesmans et al., 2008
De twee meest voorkomende mutaties in het CFTR-gen zijn die van Klasse II en Klasse III, inclusief respectievelijk de F508del mutatie en de G551D mutatie. In geval van patiënten met een Klasse II mutatie is onvoldoende CFTR in staat om het membraan te bereiken. Ongeveer 50% van de patiënten met een Klasse II mutatie hebben de F508del mutatie op beide allelen, de zogenaamde homozygoten. Voor klinische studies vormen deze patiënten een homogene groep. De overige 50% van de patiënten zijn heterozygoten die de F508del mutatie slechts op één allel dragen en een andere mutatie dragen op het tweede allel. In die gevallen heeft deze andere mutatie ook een impact op de correcte functie van CFTR. Omdat deze groep minder homogeen is, zijn klinische testen moeilijker gebleken. De F508del mutatie wordt soms een “processing” mutatie genoemd, omdat het resulteert in een defect in het CFTR-eiwit waarbij het CFTR-eiwit niet in staat is om in voldoende hoeveelheden het oppervlak van cellen te bereiken. De G551D mutatie, een Klasse III mutatie, wordt soms een “gating” mutatie genoemd, omdat het in een defect CFTR-eiwit resulteert dat wel het cel oppervlak bereikt, maar niet in staat is tot actief transport van chloride-ionen over de celmembraan. De meeste therapeutische benaderingen in ontwikkeling voor CF richten zich op de defecten veroorzaakt door één of beide van deze mutaties. Gezien de prevalentie van de F508del mutatie, zou een therapie die het effect van de F508del mutatie corrigeert, behalve voor patiënten van Klasse II mutaties, ook gebruikt kunnen worden voor combinatie therapie in heterozygote patiënten met Klasse I en Klasse III mutaties.